Expanding the druggable proteome by identifying and targeting cryptic pockets and allostery, with emphasis on viral proteins
Many proteins are currently considered to be extremely difficult to target, if not outright “undruggable.” Some appear to lack druggable pockets based on structural snapshots of what the protein typically looks like, while others have highly conserved functional sites that are difficult to drug without eliciting off-target effects by also inhibiting other proteins with the same or similar functional site.
Considering protein dynamics could greatly expand the druggable proteome by revealing features like cryptic pockets and allostery. Cryptic pockets are concavities that are absent in structural snapshots of a protein from techniques like crystallography and cryoEM but that open due to protein dynamics. Such sites can provide new opportunities for drug discovery if they either coincide with a known functional site (e.g. a cryptic pocket at a known protein-protein interaction interface could provide a means to block the proteins from binding one another) or are allosterically coupled to key functional sites.
As a specific example, we are focusing on viral proteins like Ebola VP35 (see movie above) and a number of proteins from SARS-CoV-2. Many of these proteins primarily engage in protein-protein and protein-nucliec acid interactions that are difficult to drug because the binding interfaces are large flat surfaces rather than concave pockets. Cryptic pockets and allostery could provide novel means to target these proteins, creating new opportunities for developing much needed antivirals.
Structural basis for ApoE4-induced Alzheimer’s disease
Our long-term goal is to understand the mechanism that makes polymorphism in the apolipoprotein E (ApoE) protein one of the strongest genetic risk factors for Alzheimer’s disease (AD) and to inform the development of new therapeutics for AD.
AD is the 6th leading cause of death in the USA and there are no effective treatments. Moreover, the prevalence of this age-related neurodegenerative disease is likely to increase as our population ages. Therefore, there is a great need to understand AD and develop therapeutics.
ApoE is an appealing target because this lipid transporter is one of the strongest genetic risk factors for AD (see figure below). Carriers of ApoE4 are up to 15-fold more likely to develop AD than carriers of the more common ApoE3 isoform, while ApoE2 appears to be protective against AD. Subsequent experiments have confirmed that ApoE4 plays a causal role in AD. However, the mechanism coupling ApoE and AD remains unclear.
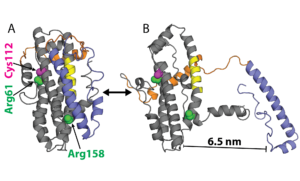
Examples of ApoE structures. A) Model of ApoE3 from NMR and B) a structure from our simulations with the N-terminal domain (NTD, gray), receptor-binding site (yellow), hinge (orange), C-terminal domain (CTD, blue), sites of polymorphism (magenta), and residues previously thought to salt bridge (green).
Neither of the substitutions that distinguish ApoE4 and ApoE2 from ApoE3 occurs in a functional site (C112R in ApoE4 and R158C in ApoE2), suggesting they indirectly impact AD risk by altering the protein’s conformational preferences. Understanding the structural differences between neurotoxic and non-toxic isoforms could enable the design of ‘structure correctors’ that combat AD by stabilizing non-toxic conformations. However, characterizing these differences remains challenging. Partial crystal structures of the different isoforms are essentially identical and the rest of the protein has largely defied structural characterization because of disordered regions and a propensity to form heterogeneous oligomers.
We are uncovering the structural determinants of ApoE-induced neurotoxicity by building and analyzing atomically-detailed Markov state models (MSMs) of neurotoxic and non-toxic variants. We are testing these models with single molecule Förster resonance energy transfer (smFRET) experiments in collaboration with Andrea Soranno and Carl Frieden.
Our models will provide insight into the connection between ApoE and AD and a foundation for precision medicine efforts, such as predicting the implications of rare ApoE isoforms for AD and designing therapeutics. Our methods will also be applicable to other proteins with disordered regions.
Adaptive sampling algorithms
Molecular simulations are an important component of all of our work. Therefore, we are also working on new methods for running these simulations as efficiently as possible. Rather than trying to run one long simulation, our main focus is on developing adaptive sampling methods. These algorithms run many independent simulations in batches. The starting points for each batch of simulations are chosen by analyzing all previous batches of simulations and determining where more simulation data would be most beneficial. Running simulations in this manner allows us to make effective use of commodity hardware and to ensure that we don’t keep simulating the same events over and over again, which is a common problem with long, conventional simulations. Deciding where to run new simulations and how to integrate the information from many independent simulations into a single, statistically rigorous model gets into lots of interesting physical chemistry and machine learning.
Extraction of valuable insight from large simulation datasets
Finally, we are drawing on information theory and deep learning to automate the extraction of valuable insights from large simulation datasets. A major theme is trying to minimize the assumptions that are built into our approaches, allowing us to be surprised by all the unexpected ways evolution has solved different problems.